Häufig gestellte Fragen (FAQ)
Chronische Entzündung
Insbesondere beim Diarrhoe-dominiertem Reizdarmsyndrom gibt es Hinweise auf Veränderungen der Darmmikrobiota, die eine Erhöhung von proinflammatorischen Proteobacteria und eine verminderte Menge von probiotischen Bakterien wie Bifidobacteria und Lactobacillus einschließen. Proteobacteria tragen toxisches Lipopolysaccharid (LPS) auf ihrer Oberfläche, das die Schleimhaut reizt. Das Fehlen schleimhautschützender Bakterien macht die Darmschleimhaut gleichzeitig weniger resilient gegenüber Toxinen und Pathogenen. Die Folge kann eine lokale Entzündung sein. Auch eine Dünndarmfehlbesiedlung (SIBO) kann Reizdarmbeschwerden verursachen. Bei 30 - 70 % der Reizdarmpatienten lässt sich eine SIBO nachweisen. Verdauungsstörungen (Maldigestion) oder eine hohe Histaminbelastung im Darm kommen ebenfalls als Ursache infrage.
Nein, im Blut persistierend hohes oder sogar ansteigendes TNF-alpha bedeutet nicht, dass die Therapie nicht wirkt. Der TNF-alpha-Blutwert ist zur Therapiekontrolle nicht geeignet.
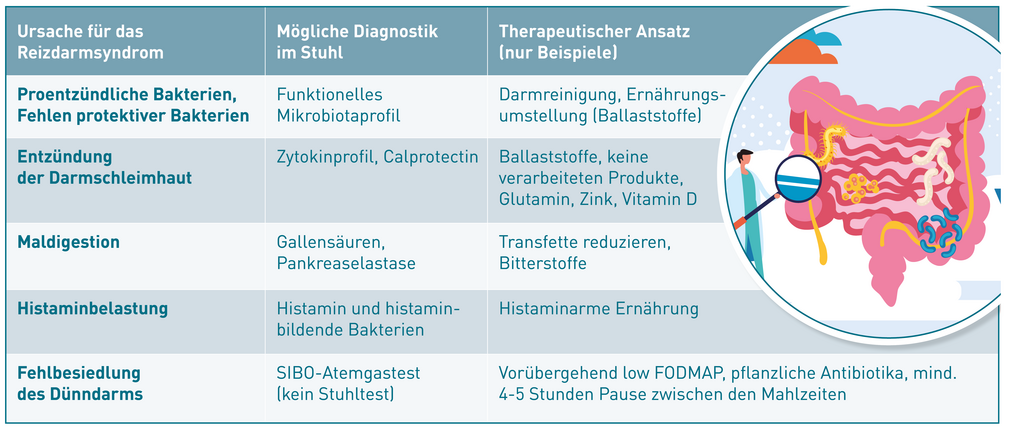
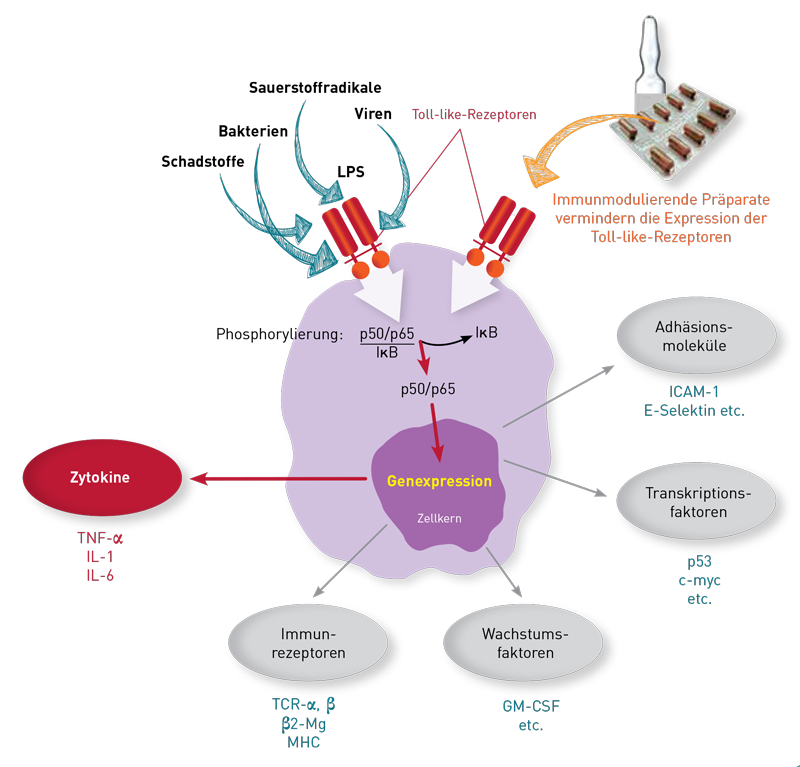
Das Entzündungszytokin TNF-alpha vermittelt seine proentzündliche Wirkung, indem es an die TNF-alpha-Rezeptoren auf seinen Zielzellen bindet. Auf diese Weise aktiviert es den Transkriptionsfaktor NF-κB, Proteasen (Caspasen) und Proteinkinasen (c-Jun N-terminale Kinase, MAP-Kinase). Diese Wirkung aktiviert die Zielzellen, was zu einer Entzündungs- und Immunantwort führt.
Die als TNF-Blocker verwendeten monoklonalen Antikörper Infliximab (Remicade®), Adalimumab (Humira®), Golimumab (Simponi®) genau wie das FAB-Fragment Certolizumab Pegol (Cimzia®) und das gentechnologisch hergestellte Protein Etanercept (Enbrel®) binden selektiv an TNF-alpha und zwar an den Teil des TNF-Moleküls, über den normalerweise die Bindung an die membrangebundenen TNF-Rezeptoren I oder II erfolgt. Keiner der genannten Antikörper hemmt die Bildung von TNF-alpha in Makrophagen, so wie das z. B. Kortison macht.
Der für die Bestimmung des TNF-Blutspiegels im Labor verwendete ELISA-Test nutzt einen "Fängerantikörper", der an eine andere Stelle des TNF-Moleküls bindet. Diese Stelle bleibt trotz der wirkungsneutralisierenden Anheftung der therapeutischen Antikörper frei, weshalb der TNF-Labortest sowohl Antikörper-gebundene als auch verbliebene freie TNF-alpha-Moleküle detektiert.
Der Grund, warum der TNF-Blutspiegel unter Therapie sogar häufig ansteigt, ist durch die fehlende negative Rückkopplung zu erklären. Physiologisch erhalten Makrophagen über ihre eigenen TNF-Rezeptoren auf der Oberfläche Signale, dass genügend TNF vorhanden ist. Da durch den therapeutischen Antikörper aber auch die rückkoppelnden TNF-Rezeptoren auf Makrophagen blockiert werden, geht der negative Feedback-Mechanismus verloren. Infolgedessen sezernieren die "falsch informierten" Makrophagen noch mehr TNF-alpha. Das zeigt aber letztlich keine entzündliche Wirkung, weil der therapeutische Antikörper auch diese Moleküle wieder neutralisiert und am TNF-Rezeptor wirkungslos macht.
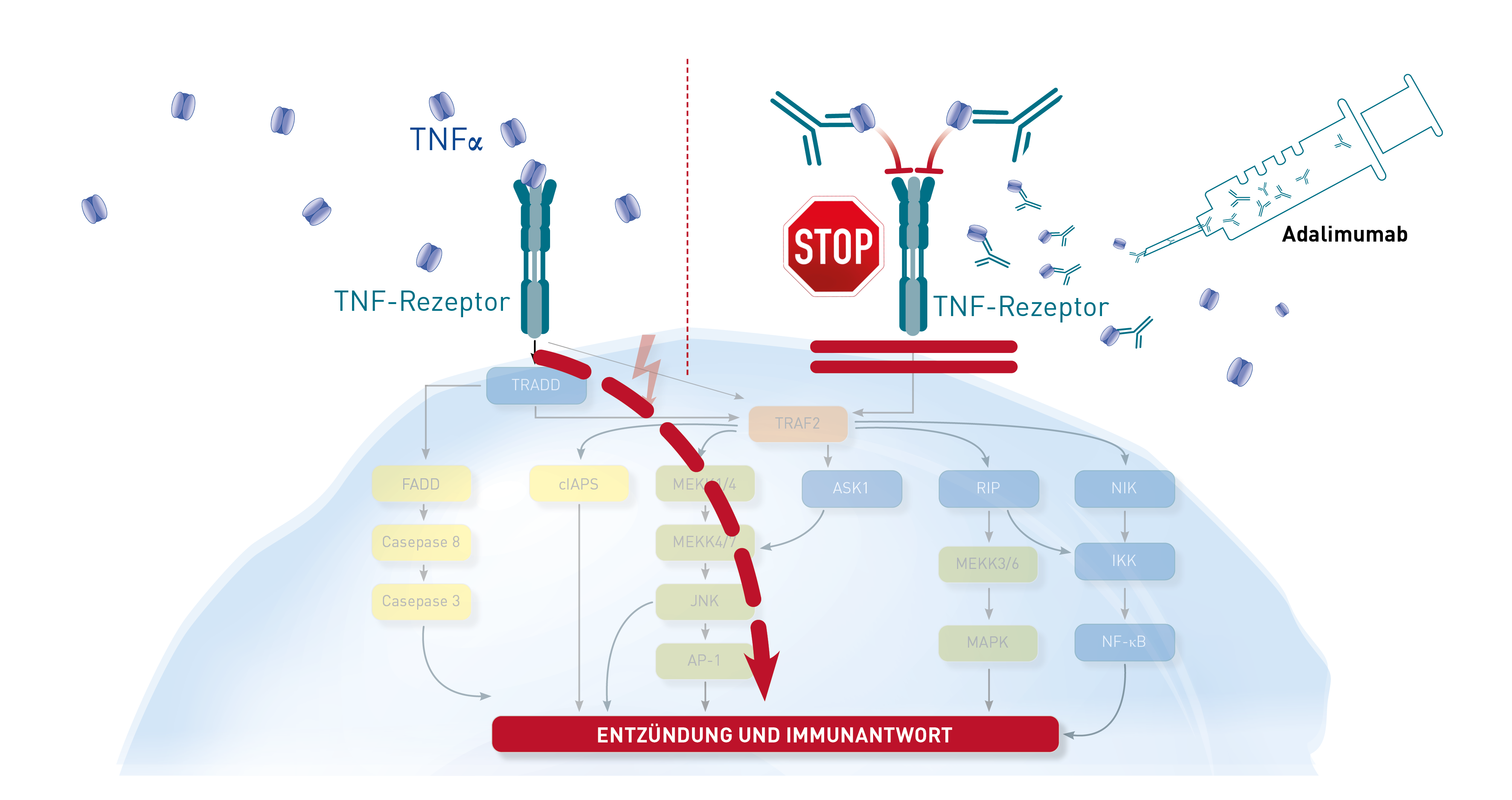
Endotoxine (Synonym: Lipopolysaccharide LPS) sind Bestandteile der Zellwand gramnegativer Bakterien. Es ist richtig, dass bei gestörter Darmpermeabilität von den Darmbakterien herrührende Endotoxine vermehrt über die Darmwand aufgenommen werden. Der Darm wird aber bekanntlich durch die Venen des Pfortadersystems drainiert. Das Pfortaderblut passiert initial die Leber, wo je nach Leberfunktion zwischen 95 und 99 % des Endotoxins schon bei der ersten Passage eliminiert werden („first pass effect“). Bis zur Blutabnahme aus der Armvene passiert das Blut noch die rechte Herzkammer, die Lunge, die linke Herzkammer und dann auch noch das gesamte arterielle Kapillarbett der Körperperipherie. Deshalb lässt der peripher-venöse Endotoxinspiegel keinen Rückschluss auf die Translokation im Darm zu. Um tatsächlich auf das im Darm translozierte Endotoxin rückzuschließen, müsste man das Blut aus der Pfortader entnehmen, was nicht möglich ist. Die Blutabnahme aus der Armvene kommt sprichwörtlich zu spät (siehe Abbildung). Ebenfalls ungeeignet zur Feststellung einer systemischen Endotoxinbelastung ist die Bestimmung der LPS-Antikörper. Die Bildung der Antikörper gegen die Endotoxinmoleküle ist variabel und wird mehr durch individuelle Antikörperbildung und -abbau beeinflusst, als durch die Menge an aufgenommenem oder zirkulierendem Endotoxin.
Für den Nachweis der systemischen Entzündung stehen heute hochsensitiv messbare Zytokine zur Verfügung (TNF-α, IL-1, IL-6). Die gestörte Darmpermeabilität kann über das Zonulin im Serum oder auch alpha-1-Antitrypsin im Stuhl erfasst werden.
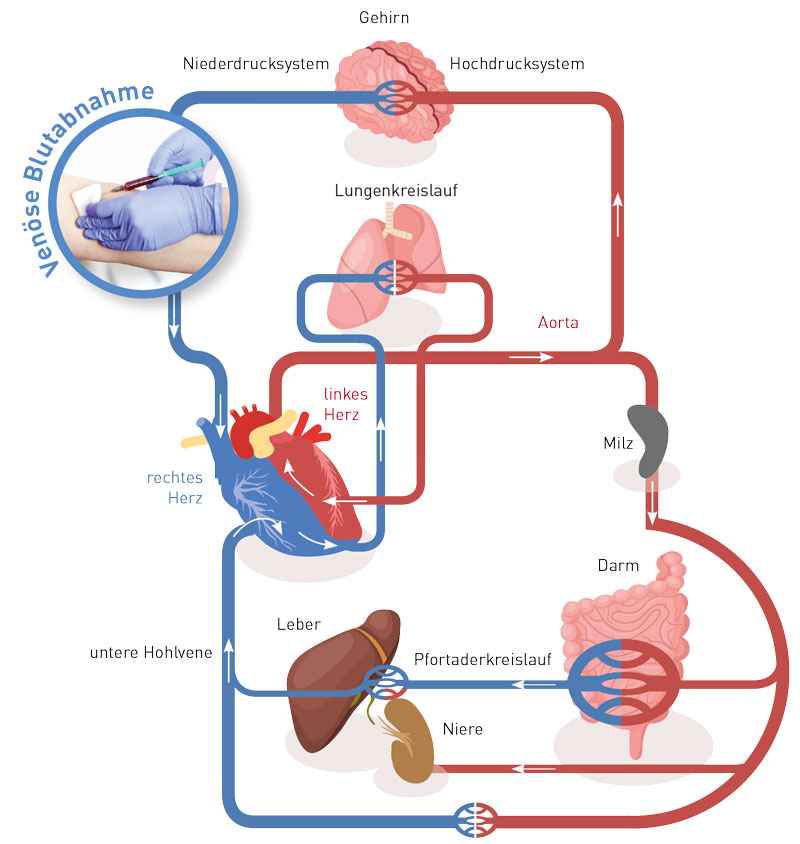
Mit Hemmtesten lässt sich die in vitro-Wirksamkeit von Immunmodulatoren auf die Freisetzung von bestimmten Entzündungszytokinen erfassen. Gut etabliert ist der TNF-α-Hemmtest, weil TNF-α am Beginn der Entzündungskaskade von Monozyten steht.
Daher lässt sich anhand des modulierten TNF-α-Spiegels unmittelbar auf die NFκB-Aktivierung der Monozyten schließen. Auch der Hemmtest für das TH2-Zytokin IL-4 ist aussagefähig, weil TH2-Lymphozyten im Blut permanent rezirkulieren.
IL-33 dagegen ist zwar in der Tat ein interessantes proentzündliches Zytokin, es eignet sich aber nicht für in vitro Hemmteste. Der Grund ist, dass IL-33 nicht oder kaum von Blutzellen sezerniert wird, sondern von Fibroblasten der Haut, Epithel- und Endothelzellen, Astrozyten im ZNS und in geringem Ausmaß auch von Promonozyten des Knochenmarkes.
Die in einer dem Patienten entnommenen Blutprobe vorhandenen Monozyten und T-Lymphozyten sezernieren IL-33 gar nicht oder nur in geringstem Ausmaß. T-Lymphozyten sind lediglich die Zielzellen von IL-33, wo es die TH2-Prägung verstärkt.
Insofern kommen wir für die Labortestung an die Zellen, die IL-33 tatsächlich sezernieren, gar nicht heran, weshalb Hemmteste, die mit peripherem Blut durchgeführt werden, für IL-33 nicht aussagefähig sind. Gleiches gilt für viele andere Zytokine.
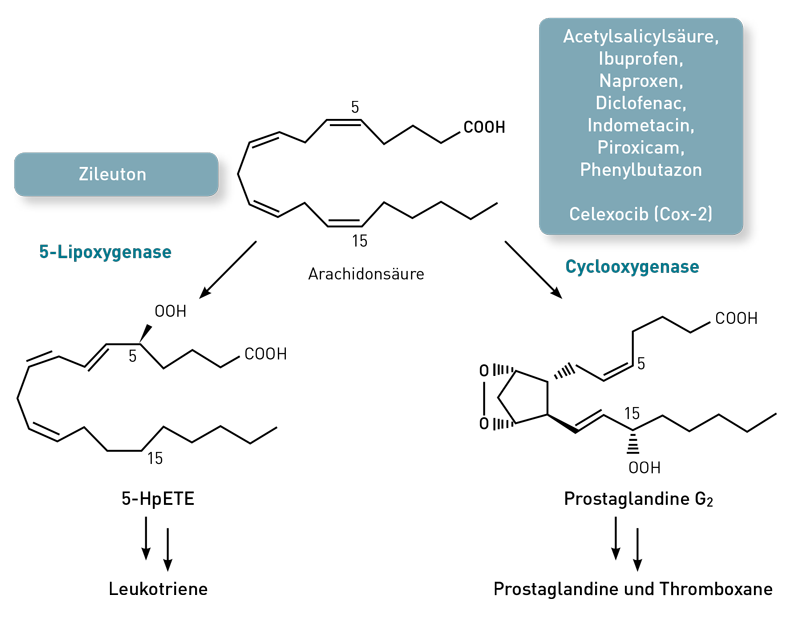
Das geht nur bedingt. Präparate wie Acetylsalicylsäure, Ibuprofen, Naproxen, Diclofenac, Indometacin, Piroxicam oder Phenylbutazon wie auch die selektiven Cox-2-Hemmer Celexocib hemmen das Enzym Cyclooxygenase (COX). Damit vermindern diese Präparate die Synthese von schmerz- und entzündungsauslösenden Prostaglandinen und Thromboxanen aus der Arachidonsäure. Der TNF-α-Hemmtest erfasst aber nicht die Prostaglandinsynthese (und somit auch nicht deren Hemmung durch Therapeutika), sondern die durch NFκB in Monozyten und Makrophagen initiierte myelomonozytäre Entzündungsreaktion. Diese läuft über TNF-α, IL-1 und IL-6. Auch die Wirksamkeit von Präparaten, welche die Lipoxygenase hemmen (LOX-Hemmer), werden im TNF-α-Hemmtest nicht erfasst.
Manchmal werden allerdings die Cyclooxygenasehemmer im TNF-α-Hemmtest bewusst mit untersucht, weil es Patienten gibt, bei denen sie die NFκB-induzierte Entzündung sogar fördern. Dieses kann in seltenen Fällen ein Therapieversagen erklären, sogar dann, wenn (was im TNF-Hemmtest aber nicht erkennbar ist) die Prostaglandinsynthese effektiv unterdrückt wird. Insofern ist also ein fehlender TNF-α-hemmender Effekt bei den NSAR und selektiven COX-2- oder LOX-Hemmern tolerabel und spricht nicht gegen die weitere Verabreichung. Bei einem TNF-α-stimulierenden Effekt sollte die Gabe allerdings kritisch überdacht werden,wenn der klinische Erfolg ausbleibt.
Interessanterweise kommt es bei einigen Patienten aber sogar zur TNF-α-Hemmung, was daraufhin deutet, dass es direkt oder indirekt (über Prostaglandineffekte auf Monozyten) doch zu einer NFκB-Hemmung durch NSAR oder COX-2-Hemmern kommen kann.
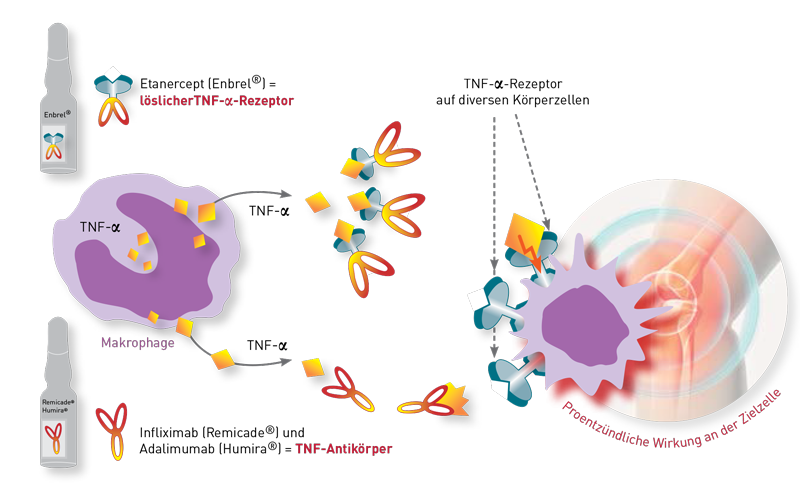
Nein. Diese Präparate bewirken ihren antientzündlichen Effekt nicht über die Hemmung der NFκB-Aktivierung in den Entzündungszellen, sondern sie vermindern die Entzündungssymptome,indem sie die Wirkung von TNF-α an den Zielzellen blockieren. Deshalb werden sie auch als TNF-α-Blocker bezeichnet.
Etanercept (u. a. Enbrel®) ist ein gentechnisch hergestelltes Fusionsprotein, welches zirkulierendes TNF-α bindet. Es fungiert somit als löslicherTNF-Rezeptor. Das von Etanercept gebundene TNF-α kann dann nicht mehr an seinen natürlichen, an der Zellmembran befindlichen Rezeptorbinden und somit keine Entzündungsprozesse im Organismus auslösen.
Ein anderes Wirkungsprinzip verfolgen die monoklonalen TNF-Antikörper Adalimumab (z. B. Humira®) und Infliximab (z. B. Remicade®). Diese Antikörper binden ebenfalls an TNF-α, allerdings nicht wie ein Schlüssel, der genau auf das Schloss passt, sondern als ein Anhängsel am TNF-Molekül. Durch die Bindung des Antikörpers an TNF-α verändert sich die Form des Zytokins mit der Folge, dass es nicht mehr in den zellständigen Rezeptor am Zielorgan passt. Der Effekt ist letztlich der gleiche. Die Auslösung oder Verstärkung der entzündlichen Reaktion durch TNF-α an den Organen wird gehemmt.
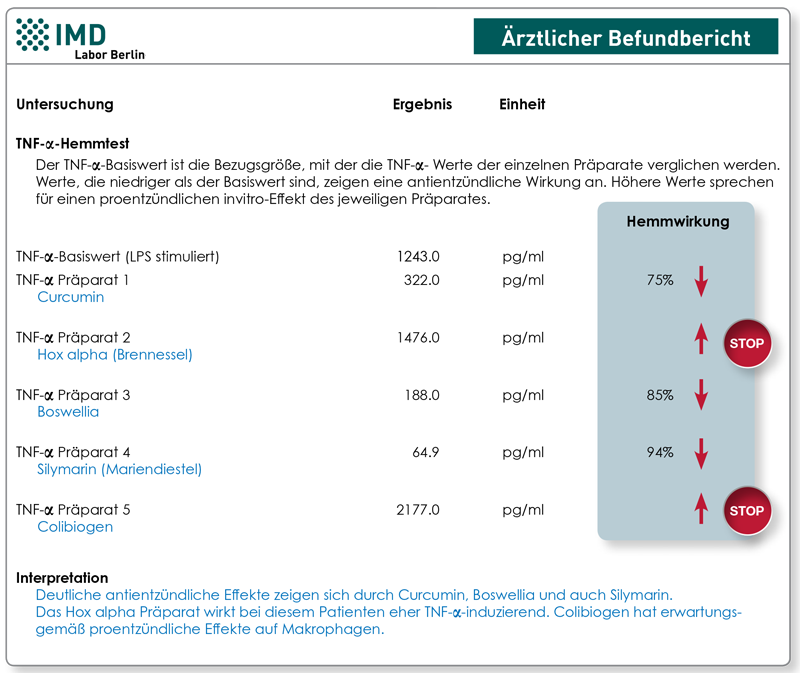
Mit dem TNF-α-Hemmtest lassen sich nur Präparate untersuchen, deren Wirkmechanismus es ist, die Aktivierung der NKκB-assoziierten Entzündungskaskarde,messbar von der Sekretion von TNF-α zu reduzieren. Dazu gehören neben Kortison auch viele Phytotherapeutika wie Curcumin, Boswellia-Präparate, Brennessel, Antioxidantien und viele weitere, oft wegen ihrer als antientzündlich bekannten Wirkung genutzten, Präparate.
Ja. TNF-α dient im Labortest nur als Markerzytokin für die Aktivierung der Monozyten und Makrophagen. Der Test misst anhand der Freisetzung des TNF-α, ob das jeweilige Präparat einen Einfluss auf die NF?B-assoziierte Entzündungskaskade hat. Die Aussage hinsichtlich der Präparatewirkung ist somit auf IL-1 oder IL-6 übertragbar. Im Vergleich zu diesen Mediatoren hat sich TNF-α als besser und sensitiver herausgestellt, was daran liegt, dass TNF-α das erste Zytokin der Kaskade darstellt und damit der geringsten posttranslationalen Modulation unterliegt. Insofern ist der TNF-α-Hemmtest bei jeder Art einer myelomonozytären Entzündung hilfreich, auch wenn diese durch ein erhöhtes CRP, IL-1 oder IL-6 nachgewiesen wurde. Der TNF-α Hemmtest ist ein globaler Entzündungshemmtest.
Keine Aussage macht der Test, ob ein Präparat antientzündlich direkt auf Lymphozyten oder Mastzellen wirkt.
Ja, man kann untersuchen, welchen Einfluss therapeutische Substanzen in vitro auf Immunzellen von
Patienten haben. Diese Vortestung mit dem TNF-α-Hemmtest ist dann sinnvoll, wenn man für eine adjuvante, antientzündliche Therapie das individuell wirksamste Präparat vor Therapiebeginn selektieren möchte.
Beim TNF-α-Hemmtest wird dem Patienten Blut abgenommen und im Labor mit Zellkulturmedium verdünnt. Anschließend wird durch Zugabe von aus bakteriellen Zellwänden gewonnenem Lipopolysaccharid (LPS) in der Zellkultur eine Entzündung induziert (Aktivierung der NFκB-assoziierten Entzündungskaskade). Diese LPS-induzierte TNF-α-Freisetzung stellt den Basiswert dar. In parallelen LPS-induzierten Ansätzen wird untersucht, welchen Einfluss zusätzlich applizierte, antientzündliche Präparate auf die NFκB-Aktivierung und die damit einhergehende Entzündung haben. Wird durch die Gabe eines Präparates der LPS-induzierte TNF-Wert niedriger als der Basiswert ohne Präparat, weist das einen antientzündlichen Effekt nach. Ist der TNF-Wert hingegen höher im Ansatz mit dem Präparat als im Ansatz ohne Präparatzugabe, dann hat das Präparat eine proentzündliche Wirkung und sollte nicht verordnet werden.
Mehr zu diesem Thema finden Sie auf unserer Diagnostikinformation Nr.: 246
Das geht meist nur bei frischen (akuten) Infektionen,nicht aber bei den häufiger zur Frage stehenden chronischen Infektionen. Bei akuten bakteriellen systemischen Infekten steigt neben CRP auch der Tumor-Nekrose-Faktor-alpha (TNF-α)und das Interleukin-6 an. Anstiege von PCT und LBP sind nur bei schweren systemischen (meist stationär behandlungsbedürftigen) Infektionen zu beobachten.In der ambulanten Versorgung sind sie entbehrlich.
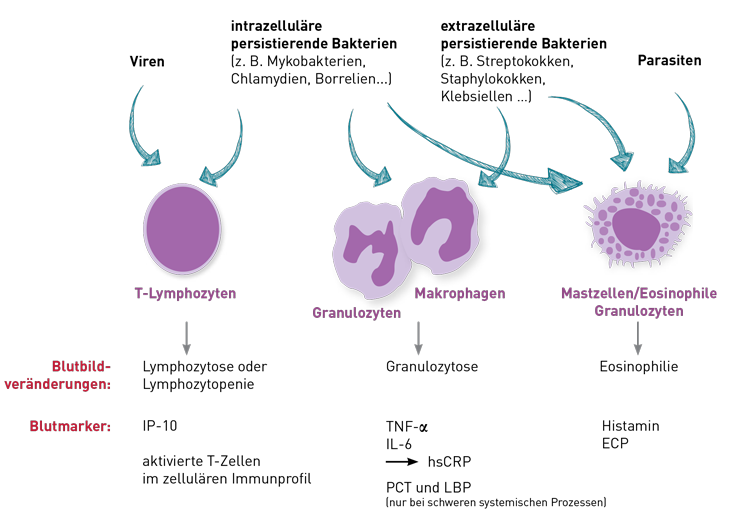
Viren induzieren dagegen eine Aktivierung von TH1-Effektorlymphozyten. Das geht mit einem Anstieg von Interferon-gamma (IFN-γ) einher. Das IFN-γ ist im Blut über den Biomarker IP-10 zu erfassen. Zytokine wie Interleukin-2 und Interleukin-12 sind im Serum kaum messbar. Ihre Bestimmung ist unnötig. Ein sensitiver Nachweis der Lymphozytenaktivierung ist über die Bestimmung aktivierter T-Lymphozyten im zellulären Immunprofil möglich.
Differenzierung zwischen bakteriellen und viralen Infektionen ist in der chronifizierten Phase über Serumzytokine nur eingeschränkt möglich, weil oft sowohl TNF-α als auch IP-10 erhöht sind. Dieses widerspiegelt den komplexen Pathomechanismus der systemischen Inflammation, wo sich Zellen untereinander aktivieren.
Erschwerend kommt hinzu, dass die Immunabwehr gegen intrazellulär persistierende Bakterien (z. B. Borrelien, Chlamydien, Mycoplasmen, Mykobakterien)eher der Immunantwort gegen Viren als der von Bakterien entspricht, da diese Immunantwort durch T-Lymphozyten getragen wird. Unsere eigenen Untersuchungen haben aber gezeigt, dass bei aktiven (chronischen) Borreliosen und Chlamydieninfekten die Zytokine im Serum häufig nicht ansteigen, d. h. normale Serumzytokine eine aktive(chronische) Infektion mit diesen Erregern nicht ausschließen.
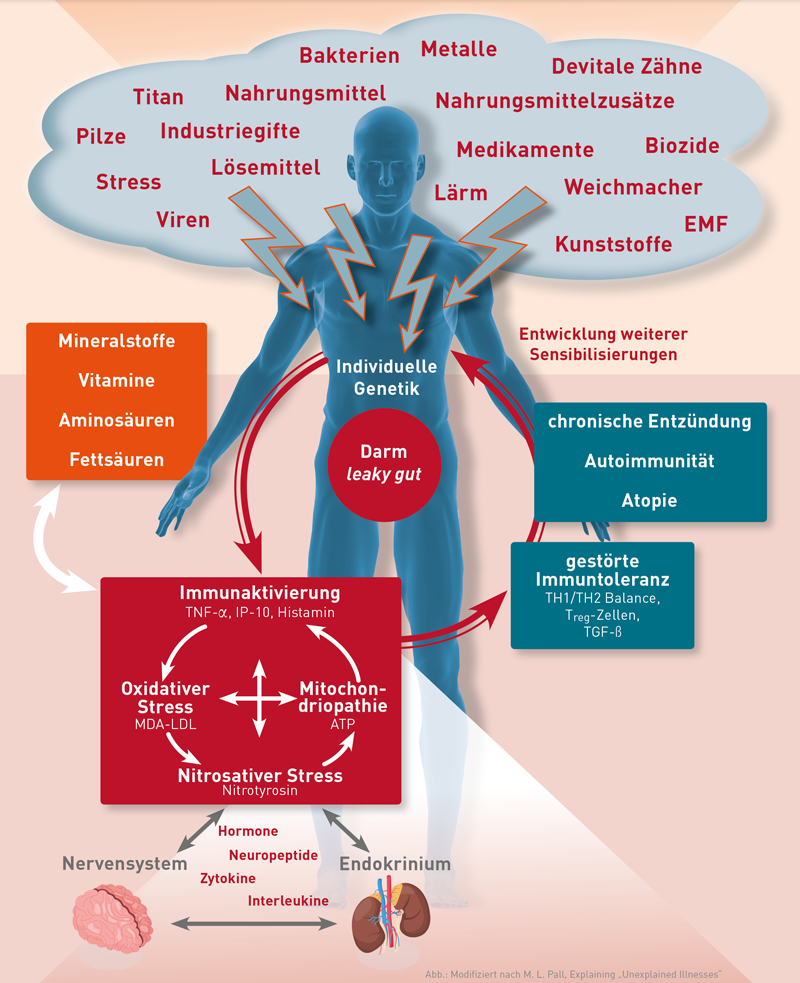
Streng genommen macht nicht nur die Immunaktivierung den Zustand der chronischen Entzündung aus, sondern auch der damit assoziierte oxidative und nitrosative Stress sowie die sekundäre Mitochondriopathie. Daher werden zumeist auch MDA-LDL, Nitrotyrosin und das in Leukozyten gemessene intrazelluläre ATP in das Profil „Chronische Entzündung“aufgenommen. Im IMD hat sich für dieses 6-Analyten-Profil die Bezeichnung „Profil Multisystemerkrankung“durchgesetzt.

Mehr zu diesem Thema finden Sie auf unserer Diagnostikinformation Nr.: 279
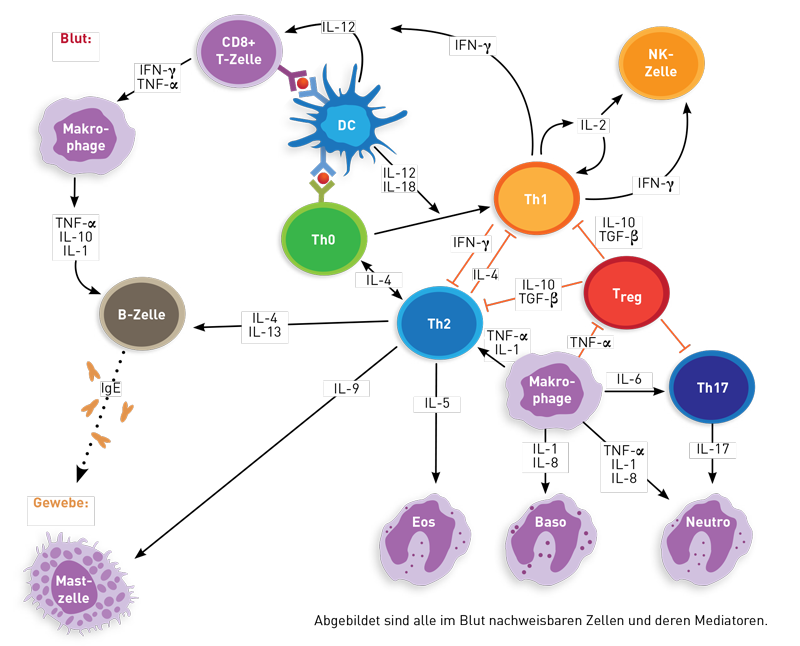
Unser Organismus verfügt über drei Entzündungssysteme, die von den unterschiedlichen Auslösern (Triggerfaktoren) differenziert aktiviert werden. So aktivieren Viren, intrazelluläre Bakterien, Tumorantigene oder auch Metalle eher die TH1-Lymphozyten (lymphozytäre Entzündung). Das unspezifische myelomonozytäre Immunsystem mit den Monozyten,Granulozyten und Makrophagen wird durch extrazelluläre Bakterien, Immunkomplexe oder auch Partikel (einschließlich Titanoxid) aktiviert.
Die Aktivierung von Mastzellen und damit die Induktion der mastzellassoziierten Entzündung erfolgt durch Allergene, aber auch durch Bakterien und zahlreiche Umweltschadstoffe. Daraus leitet sich ab, dass wir mindestens drei Entzündungsmarker messen müssen, um alle drei Entzündungssysteme sicher zu erfassen. Das sind für die myelomonozytäre Entzündung das TNF-α, für die Lymphozyten das IFN-γ (welches allerdings über seinen Biomarker IP-10 im Blut analysiert wird) sowie für Mastzellen das Histamin (im Vollblut).

Die Prävalenz chronisch verlaufender Entzündungserkrankungen nimmt in allen industrialisierten Ländern zu. Zu diesen Erkrankungen zählen Allergien, rheumatische Erkrankungen, Magen-, Darm- oder Schilddrüsenkrankheiten, Herz-Kreislauferkrankungen sowie die Parodontitis und andere chronische Infektionen. Die Fortschritte der Hochleistungsmedizin haben die Komplikationen der Erkrankungen gemindert, nicht aber deren Häufigkeit.
Warum werden chronisch entzündliche Erkrankungenhäufiger?
Im Gegensatz zur akuten Entzündung, die eine notwendige Reaktion unseres Organismus auf pathogene Eindringlinge wie Bakterien, Viren oder Pilze darstellt, ist die chronische Entzündung immer Folge einer gestörten Immuntoleranz. Ein gesundes Immunsystem kann exogene Triggerfaktoren tolerieren und eine Entzündung dem Ausmaß der tatsächlichen Bedrohung anpassen. Bei chronischen Entzündungserkrankungen handelt es sich um eine andauernde Überreaktion des Immunsystems auf zumeist harmlose Triggerfaktoren.
Was bewirken die Triggerfaktoren?
Die Abbildung zeigt die Vielfalt möglicher relevanter Auslöser einer chronischen Entzündung und soll symbolisieren, dass bei einem Patienten oft mehrere gleichzeitig einwirkende Triggerfaktoren von Bedeutung sind. In Abhängigkeit von der vorliegenden Exposition (Belastung) und den individuellen Reizschwellen (individuelle Sensibilisierung und genetische Prädispositionen?) stören die auf den Organismus einwirkenden Triggerfaktoren die Regulationstetrade aus Immunaktivierung, oxidativem und nitrosativem Stress sowie der Mitochondrienfunktion. Diese Regulationstetrade ist das Brückenglied zwischen den endogenen und exogenen Umweltfaktoren und der bei chronischen Entzündungserkrankungen gestörten Immuntoleranz.
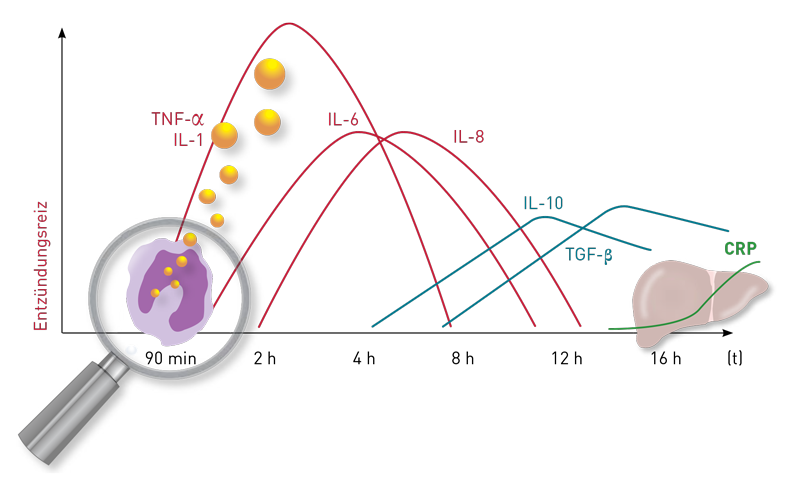
Das C-reaktive Protein (CRP) ist lediglich geeignet zum Nachweis einer akuten bzw. schwerwiegenden Entzündung, v. a. der akuten bakteriellen Infektion oder auch zum Nachweis einer Aktivitätsphase bei systemischen Autoimmunerkrankungen. CRP ist nicht geeignet zum Nachweis der Aktivierung des T-zellulären Immunsystems oder der Mastzellen, weil es in deren Mediatorkaskaden nicht vorkommt und allenfalls moderat durch Kreuzaktivierungen mit freigesetzt wird. Bei chronischen und latenten Verlaufsformen ist das CRP aber selbst zum Nachweis der myelomonozytären Entzündung zu wenig sensitiv, da es erst am Ende der Inflammationskaskade steht und nicht von den Entzündungszellen selbst, sondern von der Leber freigesetzt wird. Das betrifft auch das hoch-sensitive CRP, wo das CRP nur mit anderen Tests gemessen wird, welche den idealen Messbereich im niedrigeren Bereich haben. Zum Nachweis der latenten und chronischen myelomonozytären Entzündung ist der Tumor-Nekrose-Faktor-alpha (TNF-?) besser geeignet, da dieses Zytokin am Beginn der Entzündungskaskade steht. Allerdings sollte auch wegen der geringen Kosten das hochsensitive CRP möglichst immer mitbestimmt werden, denn es oft der Differenzierung zwischen bakteriellen und nicht-bakteriellen (viralen, allergenen) Prozessen dient.
Mehr zu diesem Thema finden Sie auf unserer Diagnostikinformation Nr.: 198
FAQ Kategorien